Anomalías en el desarrollo del Iris2
Cuanto más se estudia la linea usual o habitual del desarrollo, hay mayores posibilidades de variación y es mas difícil establecer que es normalidad.
La concepción de que es “normal”, no puede ser más que un promedio y un promedio, tendiendo a incluir mayores disparidades entre sus extremos.
Variación: Alejamiento de la teórica normalidad (calibrada desde el mayor porcentaje que existe de un determinado tipo)
Anormalidad: es una condición tan alejada de la mayoría, que no puede ser considerada una simple variación
Anomalía: es un término similar, pero menos fuerte que anormalidad.
Aberración: del desarrollo, implica que la estructura resultante no se parece a ninguna etapa “normal” en esa especie.
Detención: indica que el desarrollo simplemente se detuvo en alguna etapa, pero que no continuó por lineas anormales.
Como regla general, las estructuras mas básicas y universales aparecen primero en el desarrollo (Ontogenia), mientras que aquellas que normalmente muestran las mayores variaciones, aparecen mas tardíamente y son mas fácilmente objeto de anormalidades por ser menos estables, tanto en su desarrollo (ontogenia) como en la historia evolutiva de su especie (filogenia)
Algunas de las etapas embriológicas del desarrollo del Iris deben ser tenidas en cuenta para entender las anormalidades. Primero hay que tener en cuenta que embriológicamente hablando, las dos porciones principales: el pigmento uveal y el estroma aparecen en tiempos muy separados. La ola de la extensión mesodérmica que forma la base del estroma, se inicia alrededor de los 12mm, y a los 50-65 mm esta bien establecida, momento en el cual inicia el crecimiento del borde de la copa óptica. Las capas ectodérmicas (neuroectodermo) del Iris aparecen tardíamente y no se completan hasta el el 8avo mes, ademas el sistema vascular fetal no se atrofia o reabsorbe completamente hasta el nacimiento. Por consiguiente, es de suponer que siendo el Iris una de las últimas estructuras en desarrollarse, pueda ser objeto de anomalías menores y también de grandes anormalidades, y así es en la realidad.
En las anomalías del Iris, tanto el ectodermo como el mesodermo pueden estar implicados, juntos o en forma independiente. Por ejemplo, la persistencia de la membrana pupilar es una estructura mesodérmica normal en el desarrollo, su persistencia demuestra una detención.
De manera burda, se pueden clasificar los defectos en dos grupos:
A- Los que comprometen el espesor total del Iris (su mesodermo y su ectodermo)
B- Aquellos que comprometen un solo tipo de tejido, en forma predominante.
Defectos que comprometen el espesor total
- Ausencia del Iris ( Aniridia)
- Muescas en el Iris ( Colobomas)
- Desplazamientos y anormalidades en la forma de la pupila ( discória, corectópia, pupila lineal y policória)
- Agujeros en el Iris (dehiscencia y diastasis del Iris)
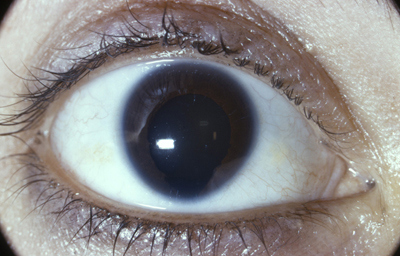
Aniridia
Es un término empleado al examen clínico, porque en todos los casos existen algunos rudimentos presentes al examen anatómico y microscópico; se asocia a mala visión, nistagmus y ausencia del reflejo foveal. (5,8)
Existen muchas teorías sobre el momento en que ocurre el error y de que tejido depende, siendo la más aceptada la teoría Ectodérmica, que postula una detención germinal de la diferenciación en la copa óptica en la etapa de los 70mm, cuando ya las capas de la retina se han diferenciado, todavía no hay fóvea, el íris ectodérmico esta presente como un estrecho reborde debajo de la unión corneoescleral, y la mayoría de los vasos cápsulo-pupilares han desaparecido. Lo que no queda claro es la razón para la ausencia casi total del iris mesodérmico, que se forma alrededor de los 48 mm. Las tendencias de la teratología hacen énfasis en la importancia de las estructuras ectodérmicas en la iniciación y control del crecimiento.
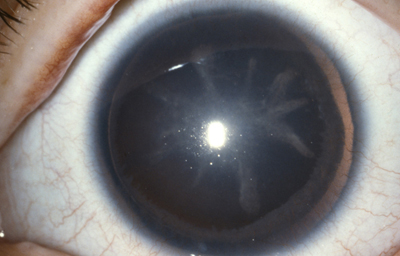
Aniridia completa, catarata y distrofia corneal de la aniridia
Archivo Dr. Francisco Barraquer
Caso de paciente con Coloboma en un ojo y Aniridia en el contralateral
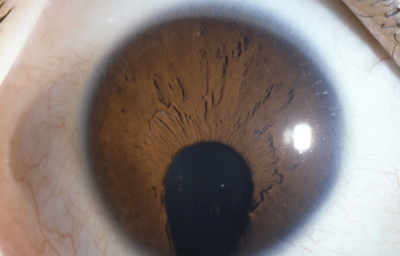
OD
Archivo Fotográfico Dr. Francisco Barraquer
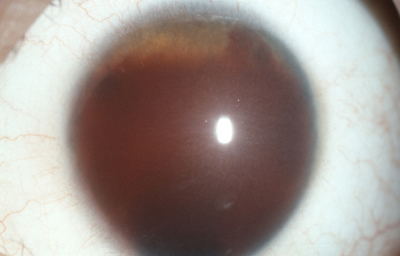
OI
Archivo Dr. Francisco Barraquer
Existen también las teorías Mesodérmicas, una de las más discutidas, es la que soporta que la falla ectodérmica es producida por persistencia indebida del mesodermo embrionario, especialmente aquel de la porción cápsulo-pupilar de la túnica vasculosa lentis. Es cierto, que en los casos típicos y atípicos de coloboma del Iris, hay evidencia clínica y patológica de persistencia indebida de uno o mas vasos cápsulo-pupilares (iridohialoideos); si se considera la Anirídia como un Coloboma Total del Iris, entonces la causa que induce la detención del desarrollo del Iris en los colobomas actuando simultáneamente en todo el círculo, causaría la Anirídia.
Actualmente se le atribuye el defecto a la región AN2 del brazo corto del cromosoma 11(11p13)que incluye el gen PAX6,(6,7) el cual ayuda a regular una cascada de otros procesos genéticos comprometidos en el desarrollo del ojo y de otras estructuras no oculares.- Es un defecto heterozigótico, es decir que solo una de las 2 copias del cromosoma 11 esta afectada.- Se transmite en forma autósomica dominante, cada descendiente tiene el 50% de posibilidad de estar afectado. Cuando ambas copias están alteradas (condición homozigótica) ocurre una condición uniforme fatal, con falla en la formación del ojo en su totalidad3 (5)
Existen mutaciones esporádicas que afectan la región WT1(Willms Tumor) adyacente a la región AN2(Aniridia) de la aniridia, causando el Nefroblastoma o tumor de Willms; las características de éste síndrome de genes adyacentes se lo conoce como WARG (T. de Willms - Aniridia - anormalidades Genitourinarias - Retraso mental) - También casos esporádicos como el Síndrome de Gillespie, causado por mutación en el gen ITPR1, ( Inosine Triphosphate Receptor with calcium channel activity) ( 4,9)
3 el gen Pax6 es considerado por algunos como el gen de control maestro para el desarrollo del ojo y cerebro; es un factor de transcripción requerido para iniciar el desarrollo del ojo en especies que van desde la mosca de la fruta ( Drosophila melanogaster) hasta los humanos. Otros aducen que se requiere de un conjunto de genes para el desarrollo del Ojo: PAX6 se expresaría como factor de transcripción cuando el ectodermo neural recibe una combinación de gradientes indicadores débiles de Sonic-SHH y fuertes de TGF-Beta
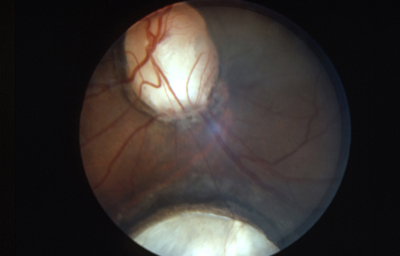
Colobomas
Desde el comienzo del estudio de la embriología del ojo, se ha hecho relación entre el cierre de la Fisura de la Copa Óptica y los Colobomas Oculares.
Se los clasifica como “Típicos” aquellos colobomas infero-nasales, y en general afectan Iris, Cuerpo Ciliar y Coroides. “Atípicos”aquellos situados en cualquier otro lugar del Iris.
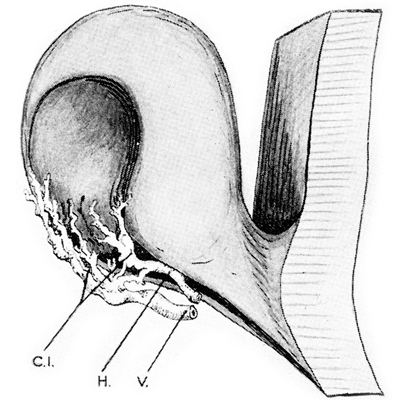
Dibujo de vaso persistente produciendo muesca en el borde de la copa
Foto tomada de: Ida Mann; The development of the Human Eye. 1969. Grune & Straton Inc N.Y
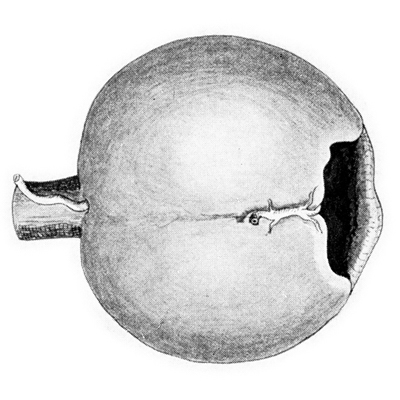
Dibujo de vaso persistente produciendo muesca en el borde de la copa
Embrión humano de 2 meses
Colobomas típicos con diferentes grados de defecto en el desarrollo
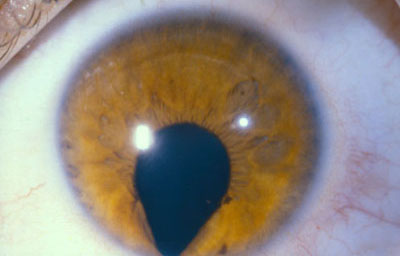
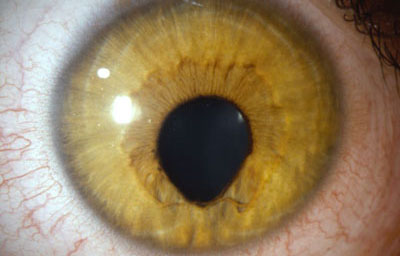
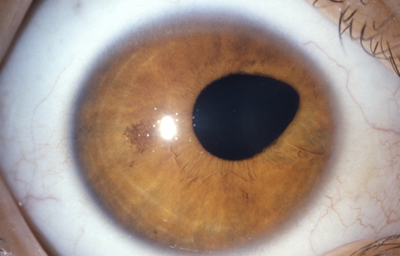
Colobomas Atípicos.
Archivo Fotográfico Dr. Francisco Barraquer
Archivo Fotográfico Dr. Francisco Barraquer
Las posibles explicaciones de su origen siguen en discusión y la pregunta es: ¿la causa primaria de los colobomas es Ectodémica o Mesodérmica? Podría tratarse de una falla localizada en una porción del margen ectodérmico de la copa óptica o bien, a una persistencia indebida del mesodermo fetal, que secundariamente inhibiría el crecimiento del ectodermo en contacto con él.
Existen múltiples argumentos relacionados, pero ninguno definitivo. actualmente se habla más de los errores genéticos que pueden producir estos defectos.
Desplazamientos y anormalidades en la forma de la pupila ( discória, corectópia y policória)
Los desplazamientos y malformaciones de la pupila forman un grupo aparte y diferente de los colobomas
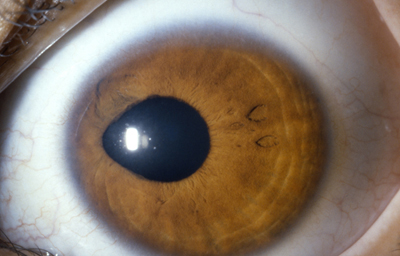
Discória: Referente de las anomalías pupilares
Archivo Fotográfico Dr. Carmen Barraquer. 1989
Corectópia: la pupila en su totalidad, esta desplazada en cualquier dirección. En muchos casos el Iris es normal excepto por su dislocación respecto al centro de la córnea. En algunos casos, se logran ver finas bandas fibrosas que determinan el desplazamiento y se cree pueden ser fibras aberrantes zonulares ( vítreo terciario); otros, consideran que pueden venir de una etapa cuando el vítreo primario ya ha desaparecido, y el vítreo secundario ( condensado formando el Haz Istmico de Druault) todavía esta unido a mesodermo del Iris.
Es muy posible que la persistencia de cualquiera de esas bandas vítreas tempranas en continuidad con el mesodermo iridiano pueda producir una deformación pupilar; su naturaleza avascular y ectodérmica explicaría su aspecto clínico.
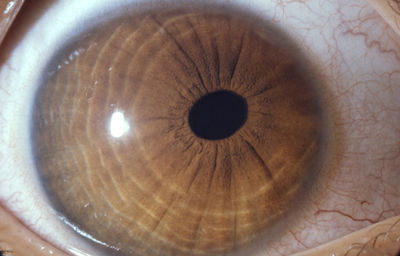
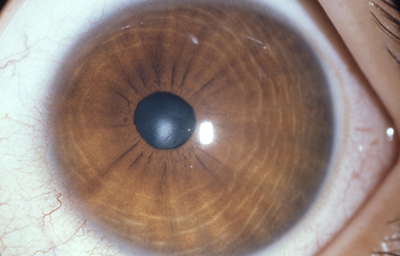
Policória: significa la presencia de varias pupilas en un Iris, es un hallazgo extremadamente raro, aclarando que el término se debe emplear solamente, para los casos en que la apertura esta rodeada por un esfínter completo; si no, es una policoria falsa.
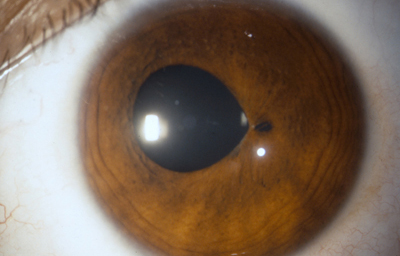
Policoria falsa en midriasis OI: sinequia en el borde pupilar temporal con banda fibrosa 3 a 4; se ve el collarete y el esfínter periféricos a la pequeña pupila, lineas de tracción superiores e inferiores hacia el puente que separa las 2 aperturas de la 1 a las 5 - dentro de apertura pequeña, parece existir un remanente de membrana pupilar.
Archivo Fotográfico Dr. Carmen Barraquer. 1983
Agujeros en el Iris (dehiscencia y diastasis del Iris)
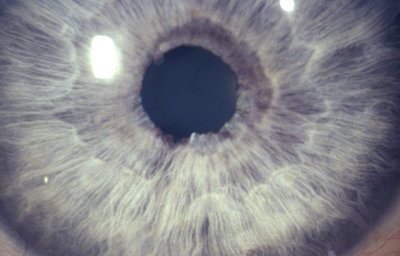
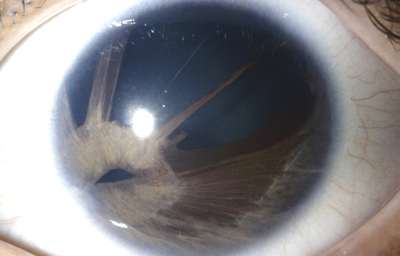
Dehiscencia: Se entiende por dehiscencia la presencia de pequeños agujeros accesorios y/o hendiduras, ademas de la verdadera pupila
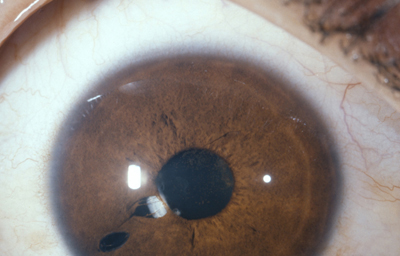
Archivo Fotográfico Dr. Francisco Barraquer
Archivo Fotográfico Dr. Francisco Barraquer
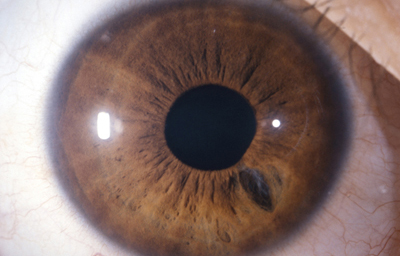
Diastasis: Significa la presencia de agujeros en la raíz del Iris parecidos a una Iridodiálisis
Diastasis del Iris: en un caso de Atrofia Esencial del Iris (Síndrome ICE)
Archivo Fotográfico Dr. Francisco Barraquer
Defectos que comprometen un solo tipo de tejido predominantemente
Defectos Ectodémicos:
- Microcoria o miosis congénita
- Ectrópion Uvea
- Entrópion Uvea
Defectos Mesodérmicos:
- Persistencia de la membrana pupilar
- Sinequias anteriores y membranas hialínas en la cámara anterior Congénitas. (Síndromes de Clivaje)
- Desordenes pigmentarios: Albinismo, Melanosis, Heterocromia y Neurocristopatías
- Anomalias del Estroma: Hiperplasia, Hipoplasia
Defectos Ectodémicos
Microcoria: Condición muy poco frecuente ( rara); es una pupila pequeña que no reacciona a la luz ni a la acomodación y no dilata mas de 1.5 mm con los midriáticos. El iris por lo demás es normal en su aspecto, sin pliegues circulares debido a ausencia del músculo dilatador parcial o totalmente, y la miosis es debida a hipereacción por la ausencia del antagonista.
Esta condición es debida a una falla en la ultima fase de diferenciación del ectodermo del Iris: al 6º mes de la gestación se inicia la aparición del músculo dilatador; en estos casos, la diferenciación del ectodermo no continua al final del 5º mes. Es un defecto familiar y hereditario. Es posible que también secundariamente el mesodermo este retrasado, explicando así, la asociación de esta malformación con la presencia de persistencia de la membrana pupilar.
El término debe reservarse para los casos con ausencia del músculo dilatador.
Ectropión congénito de la Úvea. Es una hiperplasia con eversión del epitelio pigmentario del iris, en el borde de la pupila; es raro y puede ocurrir en diferentes grados en cada caso.

A- Tres etapas en el desarrollo de un Ectropion Úvea Simple. El estroma se muestra sombreado, el pigmento ectodérmico negro.
B. Dilatación quística del seno marginal con y sin constricción por fibras mesodérmicas.
Foto tomada de: Ida Mann ; Developmental Abnormalities of the Eye. 1957. J.B.Lippincot Company. Philadelphia and Montreal
La forma mas sencilla es la simple eversión de las dos capas del epitelio pigmentario en el reborde pupilar, dando la imagen de un reborde irregular algo redundante; puede ocurrir que la eversión se extienda sobre el estroma casi hasta la periferia del Iris en un sector. No se conoce realmente si se trata de una hiperplasia ectodémica o bien de una contracción mesodérmica que hala el borde, pero no es hereditario.
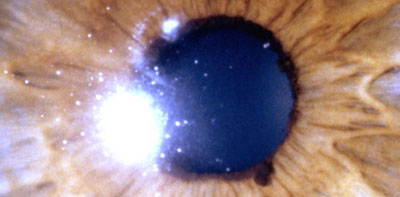
Ectropion Uvea simple
Foto tomada del archivo de los Drs. Carmen y Francisco Barraquer
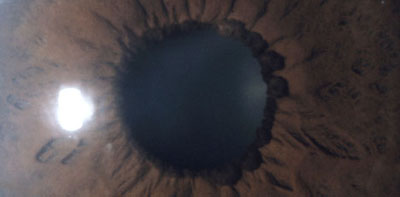
Ectropion Uvea simple
Foto tomada del archivo de los Drs. Carmen y Francisco Barraquer
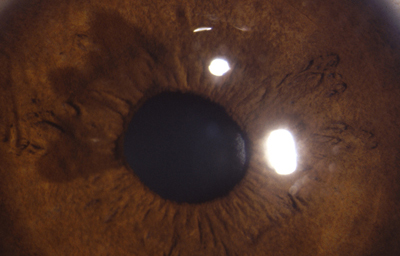
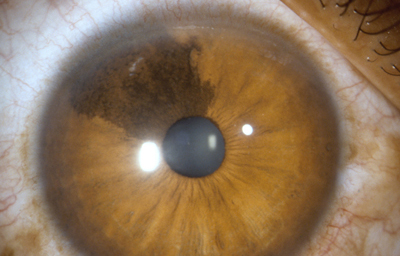
Una forma un poco mas compleja y que no siempre se denomina ectrópion, es un fruncido del borde uveal, con dilatación quística por persistencia del seno marginal; en los humanos, el seno marginal se oblitera al final del desarrollo fetal y obviamente representa la última porción de la cavidad de la vesícula óptica primaria. Cuando persiste, lo hace en forma de varios quistes de diferente tamaño en el borde, que pueden estar compuestos solo por epitelio pigmentado o tener fibras mesodérmicas de la membrana pupilar enredadas. En ocasiones estos quistes se sueltan y quedan flotando en la cámara anterior; no aumentan de tamaño ni son nocivos. Pueden aparecer con el uso de sustancias mióticas por largo tiempo.
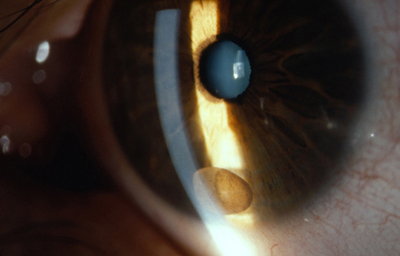
Quiste de Iris
Archivo fotográfico Dr. Carmen Barraquer
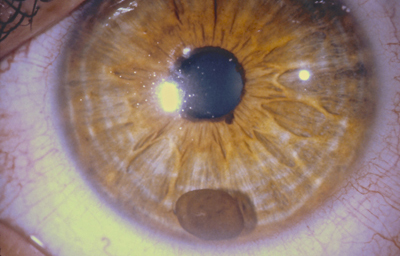
Quiste de Iris
Archivo fotográfico Dr. Carmen Barraquer
Entropion Úvea. Condición en la cual el reborde pupilar esta girado o doblado hacia la cámara anterior; es posible pero muy rara, y no hemos encontrado publicación de casos conocidos. Pudiera estar asociado a persistencia de la membrana pupilar, pero su mecanismo no es claro. Otro mecanismo mas fácil de entender sería por una contracción anormal del mesodermo, (¿inflamatoria?) en la superficie posterior del Iris.
Defectos Mesodérmicos
Persistencia de la membrana pupilar: Usualmente, el mecanismo de desaparición de la membrana pupilar es referido como atrofia.
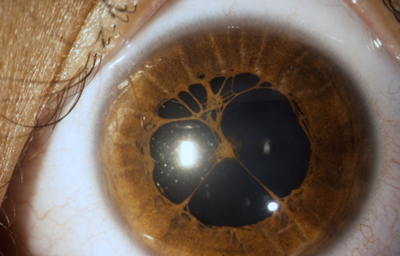
Persistencia de membrana pupilar, no hay esfínter ni collarete, detención del desarrollo?
Archivo fotográfico Dr. Carmen Barraquer
La membrana pupilar es una membrana verdadera, no una malla vascular y lo que se evidencia es un cese del crecimiento de parte de la membrana, primero las arcadas vasculares centrales (6.5 meses), después el siguiente conjunto ( 7 meses al término); gradualmente la sangre deja de circular por ellas, se rompen y desaparecen sin signos de actividad inflamatoria o mecanismo clástico, mientras tanto las porciones periféricas continuan engrosando al mismo ritmo del crecimiento del Iris. El proceso es por lo tanto una cesación del crecimiento, muerte gradual e involución o reabsorción de las células comprometidas.
Es frecuente y común ver rastros o persistencia de ella en la forma de pequeños filamentos unidos al círculo menor, en ojos por lo demás normales, y la causa es un simple fallo en la completa reabsorción y atrofia de las células comprometidas.
La causa, en los casos con persistencia de una membrana extensa es mucho más difícil de explicar. No hay evidencia de herencia. Por lo tanto el error tiene que residir en alguna condición general o local, que actúe después del 5º mes de la gestación, ya que en ese momento o un poco mas tarde, casi todos los órganos están diferenciados, pero al segmento anterior del ojo le faltan los desarrollos que están en proceso; el resto del ojo generalmente es normal.
Existen múltiples variaciones en la presentación de la persistencia de la membrana pupilar, pero la mayoría se puede incluir en ciertos grupos:
Persistencia completa: especialmente aquellos casos con circulación activa son muy raros; se ha visto más la persistencia sin circulación, en la forma de una membrana blanquecina que casi flota en el acuoso o pigmentada en casos de iris oscuro.
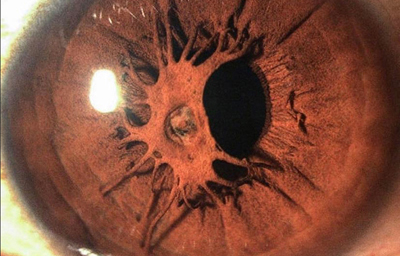
Persistencia de la membrana pupilar, debajo tiene una pupila con normal desarrollo.
Foto propiedad desconocida
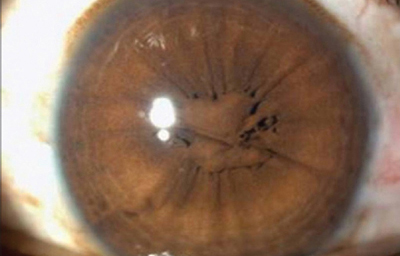
Persistencia de la membrana pupilar.
Tomada de Banigallapati et al.- Fotos tomadas de internet
Persistence incompleta: es mucho mas común y la mayoría de estos casos están dentro de los límites de la normalidad
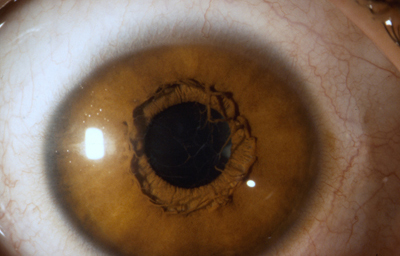
Archivo fotográfico Dr. Carmen Barraquer
a- Persistencia de uno o varios vasos únicos sin adherencias a la cápsula del cristalino, unidos al círculo menor y flotantes y/o unidos al circulo menor en sus dos extremos; según la pigmentación del iris pueden estar pigmentados o no desde el nacimiento. Esto es común.
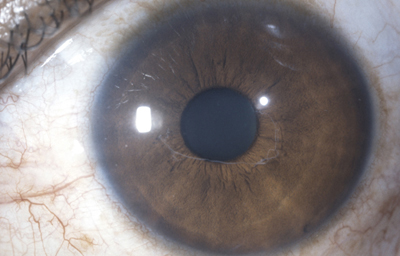
Archivo fotográfico Dr. Francisco Barraquer
b - Persistencia de vasos cápsulo-pupilares que partiendo del circulo menor, se dirigen hacia detrás del cristalino como en los casos de coloboma, pero pueden existir sin interferir en el crecimiento del Iris
c - Vasos persistentes unidos al cristalino en uno de sus extremos; parten del circulo menor y el otro extremo en contacto con el cristalino, produce una opacidad capsular anterior a veces de forma piramidal como si hubiera existido tracción sobre la cápsula.
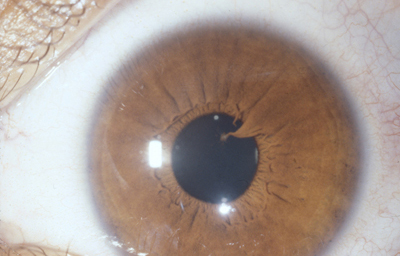
Archivo fotográfico Dr. Carmen Barraquer
d - Opacidades y hebras adherentes al cristalino pero no al Iris; pueden existir pequeñas opacidades diseminadas en el cristalino.
e - Persistencia del estroma del Iris mas allá del borde uveal de la pupila; es muy raro y se asocia a otras malformaciones oculares y sistémicas
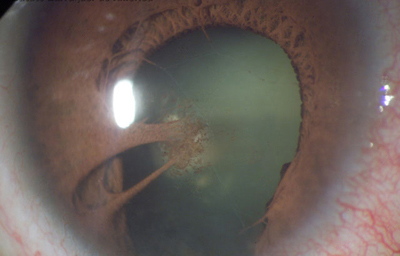
Archivo fotográfico Dr. Carmen Barraquer
f - Opacidades en la cápsula anterior sin hebras, como huellas radiales de la membrana de aspecto grisáceo tenue, en los 360º o en un sector de la cápsula, como si la membrana hubiera hecho presión sobre la cápsula antes de atrofiarse.
Sinequias anteriores y membranas hialínas en la cámara anterior Congénitas
Síndrome de clivaje de la cámara anterior. Se puede explicar como debido a error en la diferenciación del tejido post-endotelial, que en el humano (12mm) ocupa la futura cámara anterior. Ese tejido debería normalmente formar la membrana pupilar y el estroma del Iris y debería perder totalmente sus conexiones con la capa mesoblastica que forma la membrana de Descemet.
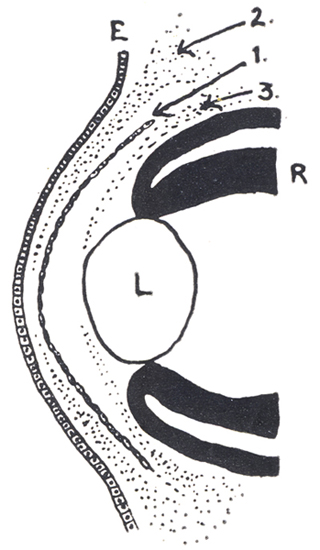
Diagrama: El crecimiento de las 3 olas sucesivas de células “mesodérmicas asociadas”, con la diferenciación de la cámara anterior:
1era - Membrana de Descemet del Endotelio
2a - Sustancia propia de la córnea
3a - El Mesodermo del Iris y la membrana pupilar
E= Epitelio corneal. R= Retina. L=Cristalino
Anomalía de Axenfeld: En 1920 Karl Theodor Axenfeld (1987-1930) caracterizó una anomalía y describió el desplazamiento anterior al Limbo corneal de la linea de Schwalbe, con filamentos (sinequias) del Iris adheridos a la linea desplazada.
El Embriotoxon Posterior es un término clínico-histológico que hace referencia la desplazamiento de la linea de Schwalbe por delante del Limbo corneoescleral.(11)
Anormalidad de Rieger: En 1935, Rieger describió una anormalidad congénita del Iris que incluye hipoplasia, corectopia y policoria. Posteriormente se encontraron anormalidades sistémicas asociadas, como defectos faciales y dentales, compromiso pituitario y anomalías umbilicales y ahora se denomina Síndrome de Rieger. (12)
La combinación de la anomalía de Axenfeld mas el síndrome de Rieger es conocido como El Síndrome de Axenfeld - Rieger ( ARS) (13)
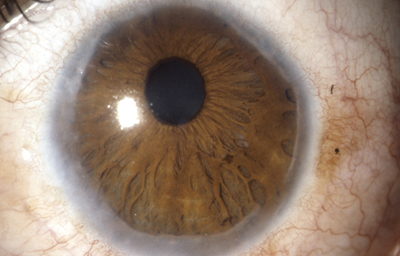
Sindrome de Axenfeld
Archivo fotográfico Dr. Francisco Barraquer
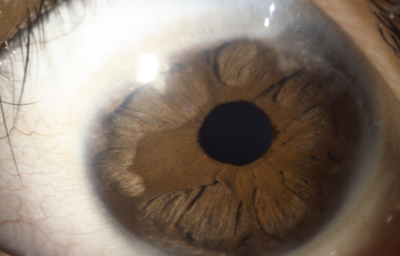
Sindrome de Rieger
Archivo fotográfico Dr. Francisco Barraquer
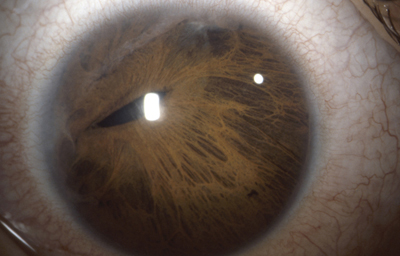
Sindrome de Axenfeld - Rieger, (ARS) Ambos ojos de un paciente
Archivo fotográfico Dr. Carmen Barraquer
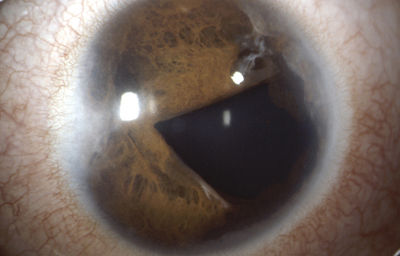
Sindrome de Axenfeld - Rieger, (ARS) Ambos ojos de un paciente
Archivo fotográfico Dr. Carmen Barraquer
Síndrome de Peters. Descrita por Albert Hans Peters (1862-1938) en 1906. Se caracteriza por ser una anormalidad en la que existe una opacidad corneal central, con ausencia de membrana de Descemet y Endotelio en la zona de la opacidad y adherencias iridocorneales con patrón variable a la zona corneal afectada; muchos casos se asocian con Glaucoma congénito, Aniridia y Microcórnea. Se considera que pudo existir un retraso en la separación del cristalino de la placa ectodermica superficial que le dio origen. En el S. de Peters se describen tipo 1, el tipo 2 y el tipo Peters Plus, en el cual existen alteraciones en el cristalino, tiende a ser bilateral y se asocia con anormalidades sistémicas como retraso del desarrollo, defectos cardiovasculares, mala audición, defectos en el sistema nervioso central, defectos genitourinarios y gastrointestinales.
El compromiso corneal lo diferencia del Síndrome de Axenfeld-Rieger
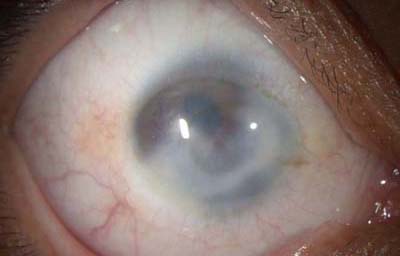
Sindrome de Peters
Archivo fotográfico Dr. Carmen Barraquer
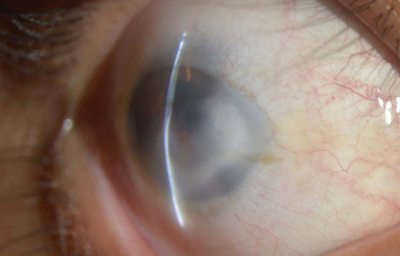
Sindrome de Peters
Archivo fotográfico Dr. Carmen Barraquer
Genéticamente el Síndrome de Clivaje es un grupo de anormalidades heterogéneo, como resultado de mutaciones por lo menos en 4 locus genéticos diferentes. Han sido asociadas mutaciones en PITX2 en 4q25, FOXC1 en 6p25, PAX6 en 11p13, y FOXO1A en 13q14 y FOXO1A en 13q1. también se han reportado casos del síndrome de Axenfeld-Rieger con mutaciones en CYP1B1.En general son esporádicas, pero se han reportado casos con herencia autosómica dominante y recesiva. (14)
Estos genes, actuarían en concierto en la regulación de la diferenciación de las células mesenquimales provenientes de la cresta neural, que dan origen a diferentes tejidos en el segmento anterior del globo ocular. El desarrollo, parece ser muy sensitivo a la dosificación genética; la variación del nivel normal de actividad de los factores de transcripción, sería el causante de las anomalías en el segmento anterior.
Síndromes Adquiridos
Síndrome Endotelio-Irido-corneal, = ICE (Iridocorneal Endothelial Syndrome)
Grupo de alteraciones caracterizado por anomalias proliferativas del endotelio corneal, anormalidades en el Iris y del ángulo de la cámara anterior. Clínicamente se observa edema de la córnea, atrofia del Iris, anomalías pupilares y glaucoma.
Abarca tres variantes 1) El Sindrome de Chandler.- 2) La Atrofia Esencial del Iris.-3) El Síndrome de Cogan-Reese ( Nevus del Iris)
Patológicamente aparece como una enfermedad adquirida en la que las células endoteliales adquieren características de células epitelioides, con habilidad migratoria, tonofibrillas y desmosomas. Se considera que el aspecto del “Endotelio-martillado de color plata” es un signo diagnóstico.
Tipicamente es unilateral, progresivo, con predominancia femenina, se manifiesta al inicio de la vida adulta (20-50 años) y no se asocia a malformaciones sistémicas.
Etiología desconocida; se plantea posibilidad de relación con el virus del Herpes.
El Sindrome de Chandler. Se llama así, cuando los cambios patológicos están limitados a la superficie interna de la córnea con alteración de la bomba endotelial, lo que produce edema de córnea. Si el endotelio anómalo invade el ángulo, se desarrolla glaucoma.
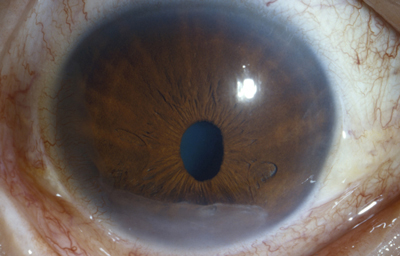
Sindrome de Chandler
Archivo fotográfico Dr. Francisco Barraquer
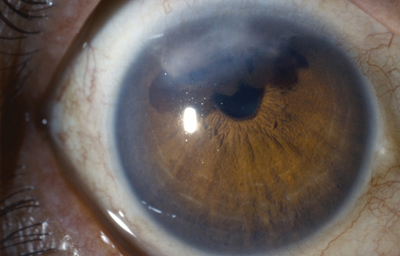
Sindrome de Chandler
Archivo fotográfico Dr. Francisco Barraquer
La Atrofia Esencial del Iris. Se produce cuando el endotelio anormal prolifera sobre la superficie del Iris, con subsecuente formación de una membrana contráctil que produce corectopia, atrofia del Iris y policoria falsa; si el endotelio anormal prolifera sobre el ángulo de la cámara anterior genera sinequias y glaucoma.
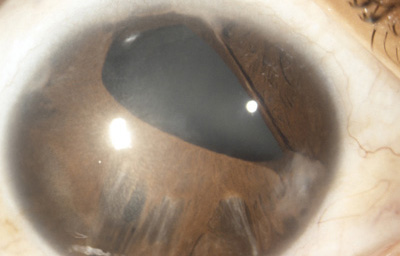
Atrofia Esencial del Iris - OD de paciente femenina de 36 años
Archivo fotográfico Dr. Carmen Barraquer
El Síndrome de Cogan-Reese ( Nevus del Iris). Se manifiesta por numerosos nódulos iridianos pigmentados, causados por contracción de membranas endoteliales anómalas, sobre la superficie del Iris- produce Glaucoma.
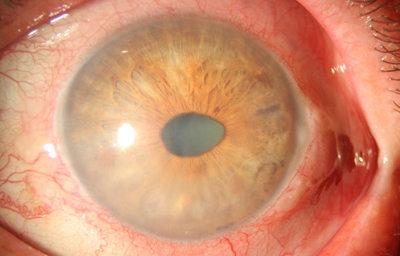
Síndrome de Cogan-Reese
OD : con discoria, anisocoria y edema corneal
Archivo fotográfico Dr. Francisco Barraquer
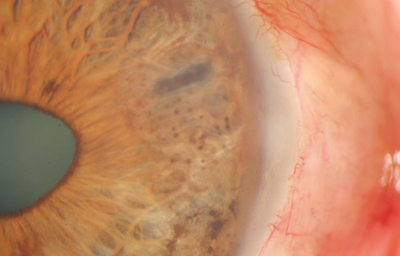
Síndrome de Cogan-Reese
Detalle de Nódulos iridianos pigmentados
Archivo fotográfico Dr. Francisco Barraquer
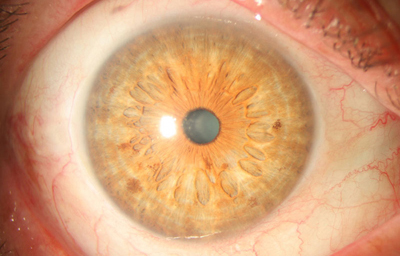
Síndrome de Cogan-Reese
El OI sin alteraciones
Archivo fotográfico Dr. Francisco Barraquer
Diagnóstico diferencial con otros síndromes ICE, Melanocitoma del Iris, Neurofibromatosis, Melanoma, Glaucoma neovascular
Defectos Pigmentarios: Albinismo, Piebaldismo, Melanosis, Heterocromia, Neurocristopatías
Albinismo: Hereditario en forma recesiva, en los humanos tienen ausencia completa de pigmento mesodermico en todo el cuerpo y falla parcial de pigmento ectodérmico.
Albinismo Oculocutáneo tipo I, autosómico recesivo debido a el gen para la Tirosinasa ( gen OCAI) cuando no funciona bien.
Albinismo Oculocutáneo tipo II en el cual se ha codificado la molécula P (OCA2) que es una proteína integrada en la membrana, dedicada a transportar moléculas pequeñas, específicamente Tirosina ( precursor de la Melanina)
Albinismo Ocular tipo 1 (OA1): es el más común, se hereda en forma recesiva ligado a X en el que no hay epitelio pigmentado en la retina, pero la piel es normal- Tienen nistagmus Gen (GPR1).
Albinismo Ocular tipo 2: Asociado a discromatopsias y a nictalopia Genes (CAC y NA1F). localizado en el brazo largo (q) del cromosoma 15
Se menciona un tercer tipo asociado a Sordera, que pudiera ser el mismo OA1
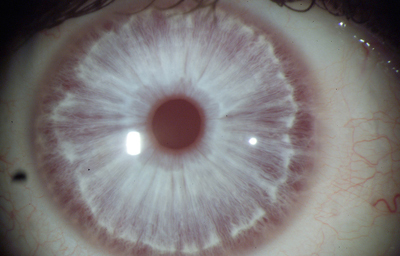
Archivo fotográfico Instituto Barraquer de América
Piebaldismo: ( Neurocristopatía)(Neuropatía de la Cresta Neural) No confundir con “albinismo”; es un defecto en el desarrollo de los melanocitos, en el que pueden producir pigmento pero esa función esta detenida o apagada. Tienen Epitelio pigmentario normal y visión normal. Es un defecto autosómico dominante. Manchas blancas en piel.
Los caballos y vacas “Pintos” al igual que otros animales con parches de color en la piel son Piebaldos.(14)
Ver el Síndrome de Waardenburg
Melanocitosis Ocular unilateral ( Melanosis ocular) Es una heterocromía unilateral con hiperpigmentación, puede verse en los nevus de Iris; cuando la hiperpigmentación compromete la esclera y la piel periocular, se la conoce como nevus de Ota; se debe hacer diagnóstico diferencial con Melanocitoma y con Melanoma Difuso del Iris
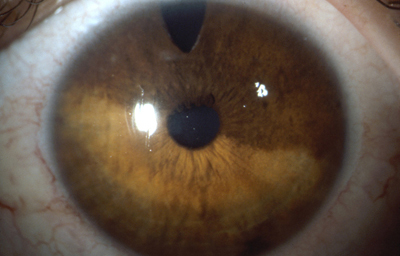
Melanocitosis Ocular
Archivo fotográfico Dr. Carmen Barraquer.
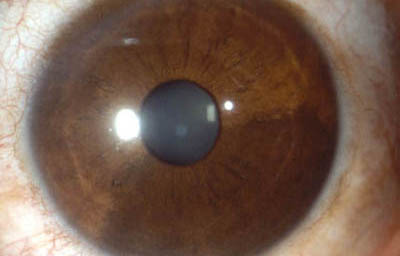
Melanocitosis Ocular
Archivo fotográfico Dr. Francisco Barraquer.
Nevus de Ota
Archivo fotográfico Instituto Barraquer de América.
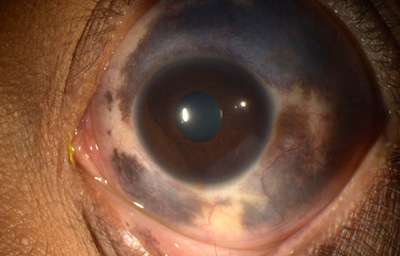
Nevus de Ota
Archivo fotográfico Instituto Barraquer de América.
Defectos pigmentarios unilateral: ( con Hipopigmentación)
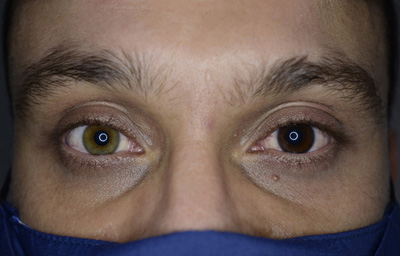
Heterocromia Simple
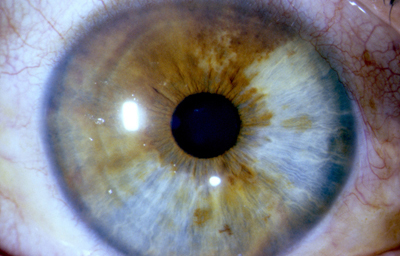
Heterocromia con Paralisis simpática:
Síndrome de Claude Bernard - Horner: hipopigmentación del Iris, anisocoria (miosis), ptosis, anhidrohidrosis, enoftalmos, defecto refractivo.
Puede ser congénito o adquirido y es producido por lesión del tronco simpático ipsilateral. La heterocromia aparece en los casos congénitos.
Aunque en la mayoría de los casos es una lesión menor, puede reflejar enfermedades como el tumor de Pancoast ( tumor en el apex pulmonar) o una dilatación venosa tiroidocervical
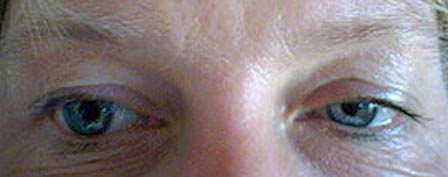
Heterocromia con Paralisis simpática
Foto tomada de Internet
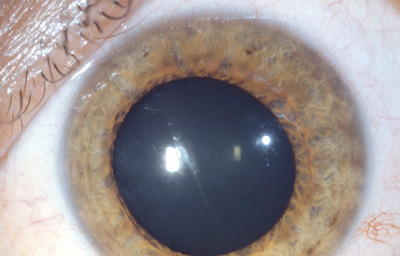
Síndrome Uveitis heterocrómica de FuchsUveitis de etiología indeterminada, se caracteriza por iridociclitis crónica unilateral con despigmentación del iris afectado, y precipitados queráticos; a largo plazo catarata y glaucoma. Es poco sintomática y no se han encontrado enfermedades sistémicas asociadas
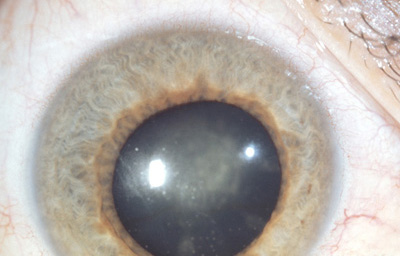
Sindrome Uveitis heterocrómica de Fuchs
OD Iris claro en midriasis, con precipitados queráticos en el endotelio corneal y catarata.
Archivo fotográfico Dr. Carmen Barraquer.
Sindrome Uveitis heterocrómica de Fuchs
OI Ojo Izquierdo sano. Iris mas oscuro, en midriasis, cristalino trasparente.
Archivo fotográfico Dr. Carmen Barraquer.
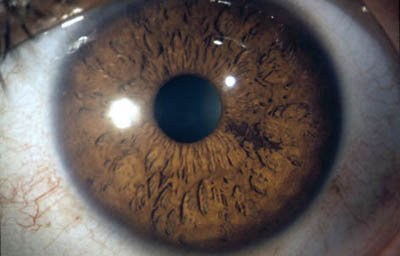
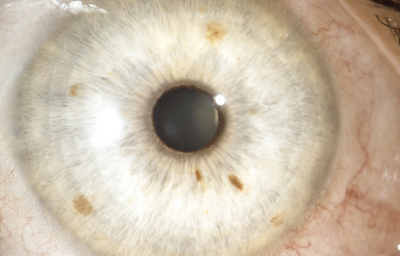
Síndrome de Waardenburg (Bilateral) PAX3 (Neurocristopatia) (22,23)Es un grupo con unas condiciones genéticas raras por lo general de herencia autosomica dominante, es una forma de albinismo irregular, fluctuante; tienen epitelio pigmentario normal y visión normal; sinembargo en el tipo II y IV hay también herencia autosomica recesiva.- Se describen 4 tipos en este síndrome.
El tipo 1 se caracteriza por Telecanto, cierto grado de Hipoacusia congénita y deficiencias pigmentarias en piel con manchas blancas, Mechon blanco en el pelo y deficiente pigmentación del Iris bilateral. Mutación del PAX3
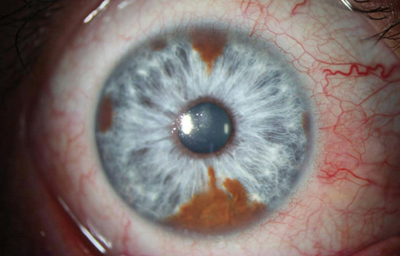
OD
Archivo fotográfico Dr. Francisco Barraquer.
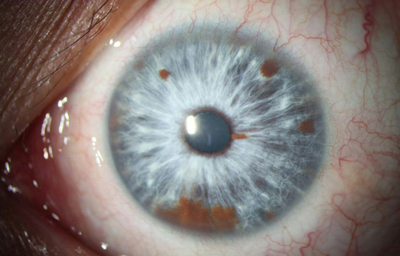
OI
Archivo fotográfico Dr. Francisco Barraquer.
El tipo 2: es el mas frecuente siendo raro, Pueden tener mechón blanco en el cuero cabelludo, manchas blancas en la piel y heterocromia del iris por hipopigmentación bilateral. Mutación del MITF( factor de transcripción asociado a microoftalmia). Aquí se describe también, el Síndrome de Tietz,(descrito en 1963 por Walter Tietz) semejante al tipo 2 con Hipoacusia neurosensorial e Hipopigmentación generalizada de la piel a diferencia de los parches en el Waardenburg. Los ojos tambien son afectados, con Iris claro e hipopigmentación en el epitelio pigmentario de la retina. Autosómico dominante ( Mutación MITF)4
Tipo 3 - Es una mutación más severa del tipo 1, en el cual las manos y brazos también tienen malformaciones: contracturas permanentes y dedos fusionados. Mutación del PAX3
Tipo 4: Tiene enfermedad de de Hirschsprung en el que hay ausencia de inervación intestinal.Mutación del SOX10
4 El factor de transcripción asociado con microftalmia (MITF) es un factor de transcripción con un dominio hélice-bucle-hélice básico de cremallera de leucina, implicado en el desarrollo de melanocitos1 y osteoclastos.
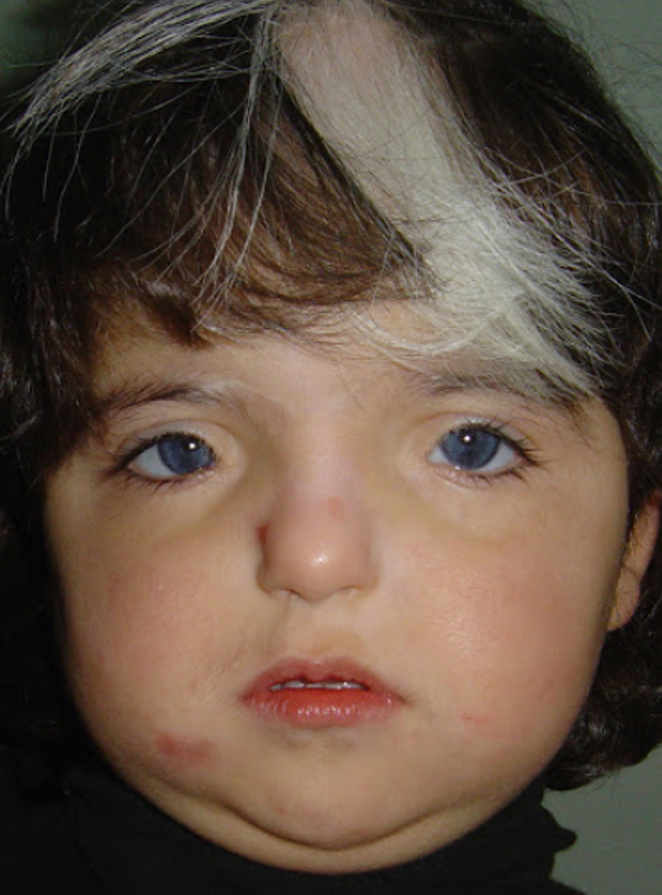
OI
Foto tomada de internet
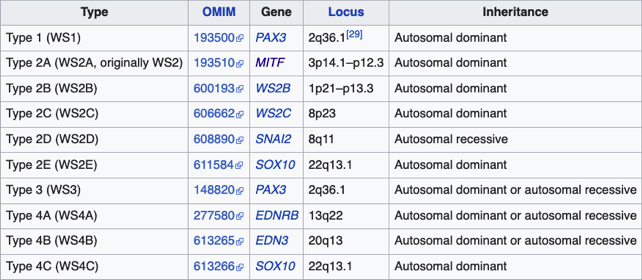
Anomalias del Estroma: Hiperplasia, Hipoplasia
Hiperplasia
Archivo fotográfico Dr. Francisco Barraquer.
Hipoplasia
Archivo fotográfico Dr. Francisco Barraquer.